Image by Robin Skånberg
VIAMD (Visual Interactive Analysis of Molecular Dynamics)
InfraVis Collaborators
InfraVis Application Expert
Robin Skånberg (KTH)
InfraVis Node Coordinator
Björn Thuresson (KTH)
Tools & Skills
C/C++, OpenGL, Molecular Dynamics
Keywords
Molecular Dynamics, Molecular Visualization, Trajectory Analysis, Interactive Visualization, Scientific Computing
VIAMD (Visual Interactive Analysis of Molecular Dynamics) originated as a research project developed during a PhD in computational chemistry and scientific visualization. The initial motivation came from the practical challenges of analyzing molecular dynamics simulations in day-to-day research, where understanding simulation output often required moving between multiple disconnected tools for visualization, scripting-based analysis, and post-processing. In typical workflows, molecular trajectories are inspected in a visualization program, exported for analysis in separate environments, and then re-imported or re-plotted to interpret results. This separation makes it difficult to iteratively refine hypotheses or directly connect observed structural behavior with quantitative measures.
VIAMD was developed to explore a more integrated approach, where interactive 3D visualization and quantitative analysis coexist within the same environment. Early versions of the system were built to support exploratory analysis of molecular trajectories during research work, gradually evolving into a more general platform for interactive molecular data analysis. The project grew out of the need for tighter coupling between simulation data, structural interpretation, and derived physical properties, with a particular focus on enabling real-time feedback between user-defined analyses and molecular visualization. Over the course of the PhD, VIAMD evolved from a research prototype into a more general tool for interactive scientific exploration of molecular dynamics datasets, driven by recurring analysis needs in computational chemistry workflows.
Target audience and interactions
VIAMD is intended for researchers working with molecular simulations who need to explore and interpret large trajectory datasets interactively. Users can load molecular dynamics trajectories, inspect molecular structures in 3D, and simultaneously investigate computed properties through linked visual analysis views. Instead of treating visualization and analysis as separate workflows, VIAMD allows researchers to directly connect molecular structures, temporal behavior, and statistical properties. The interactive environment supports exploratory analysis, where users can move between observing molecular motion and investigating quantitative features of the simulation data.
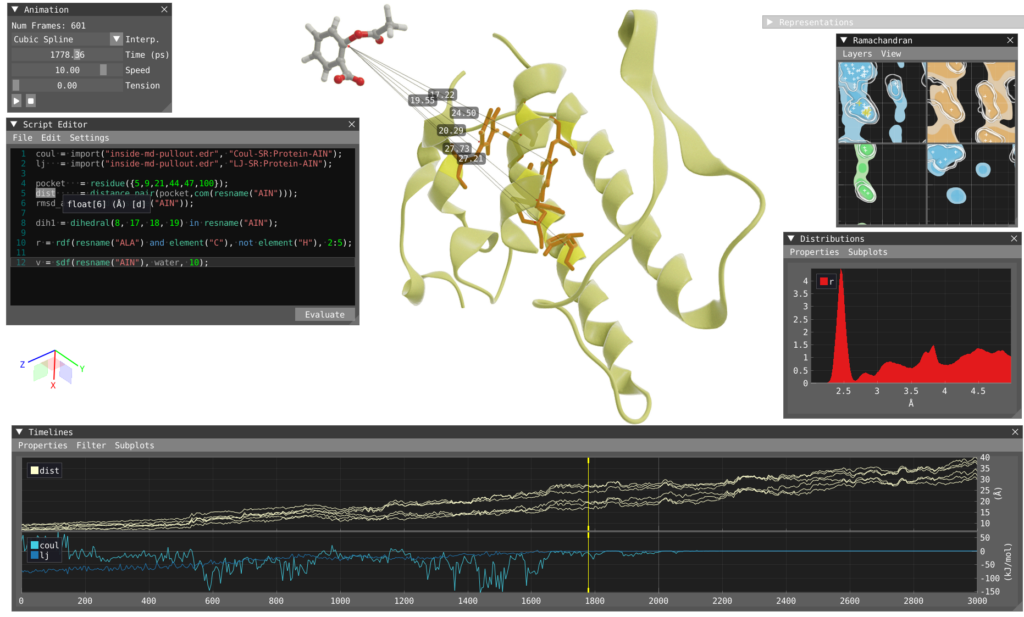
The visualization shows a complex formed between group II phospholipase A2 and aspirin.
Image by Robin Skånberg.
Interactive analysis of molecular simulation data
A central feature of VIAMD is the ability to define and evaluate custom properties through a scripting interface. This allows researchers to create analyses specific to their scientific questions without requiring changes to the core application.
Computed properties can be explored through different visualization components, including:
- Timeline-based analysis of molecular properties over trajectories
- Distribution views for statistical evaluation
- Density-based representations of molecular behavior
- Interactive linking between analysis results and molecular structures
This enables researchers to identify patterns and events in simulations while maintaining direct visual context.
Technical details
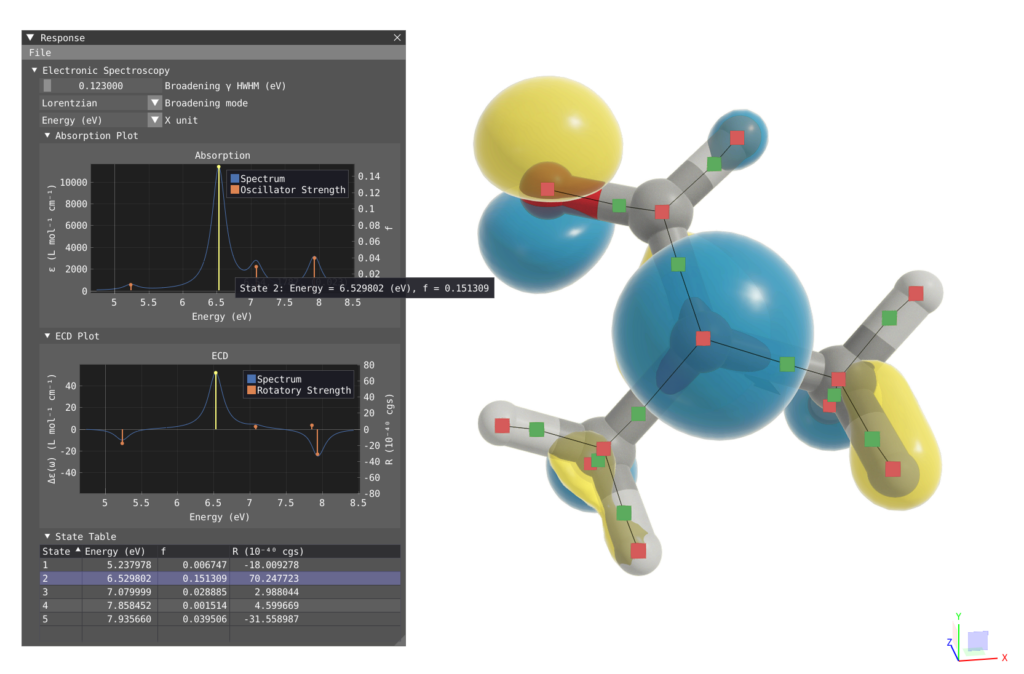
VIAMD is implemented as a high-performance desktop application optimized for interactive visualization of molecular simulation data. The software uses a multi-threaded architecture to handle large datasets while maintaining responsive interaction. The current implementation primarily supports molecular dynamics formats used in simulation packages such as GROMACS, with ongoing development focused on expanding data support and integration with additional computational workflows. Recent development includes extending VIAMD towards visualization and interactive analysis of electronic structure calculations, including integration with data from the VeloxChem software ecosystem.
Impact
By combining visualization, computation, and analysis into one interactive workflow, VIAMD aims to reduce the gap between molecular simulation data and scientific interpretation. The project provides researchers with a flexible environment for exploring molecular behavior and supports the development of new approaches for analysing complex simulation datasets. VIAMD is available as open-source software under the MIT license, with documentation, tutorials, and examples available for users and developers.
Code: https://github.com/scanberg/viamd
Documentation: https://github.com/scanberg/viamd/wiki
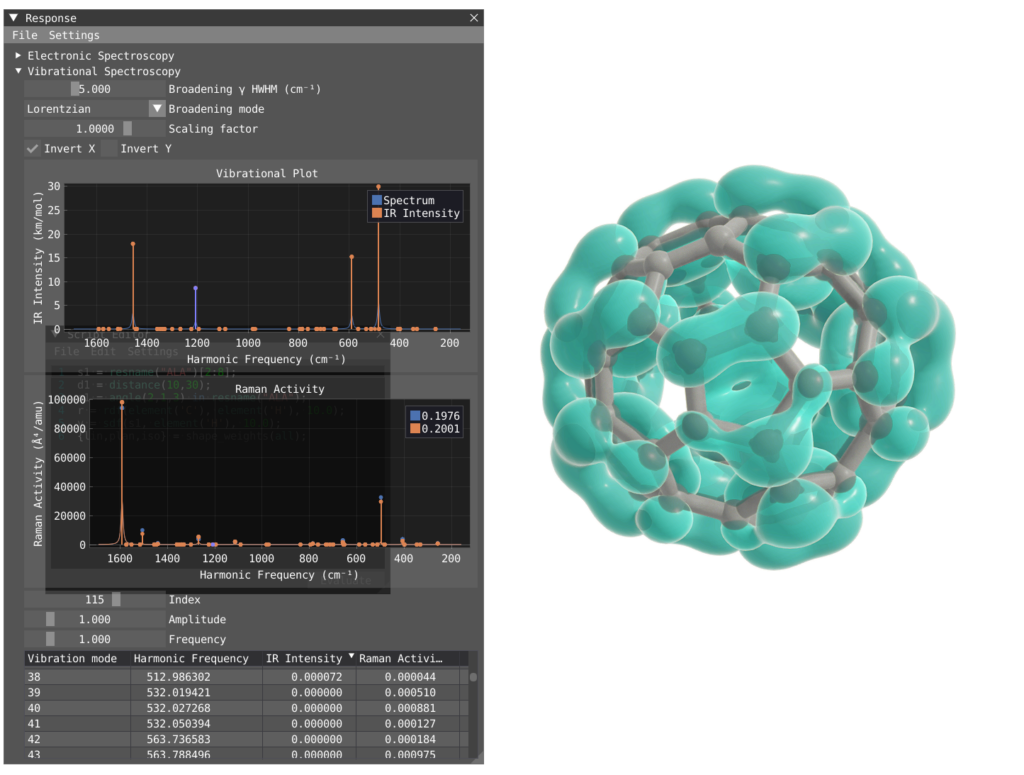
Image by Robin Skånberg.